UCSC Genome Browser Tutorial
1. Overview of UCSC genome browser
Section titled “1. Overview of UCSC genome browser”The UCSC Genome Browser provides a rapid and reliable display of any requested genomic region at any scale. It supports a wide range of genome assemblies across various species (eg. human GRCh38 and mouse mm10) and includes dozens of aligned annotation tracks.
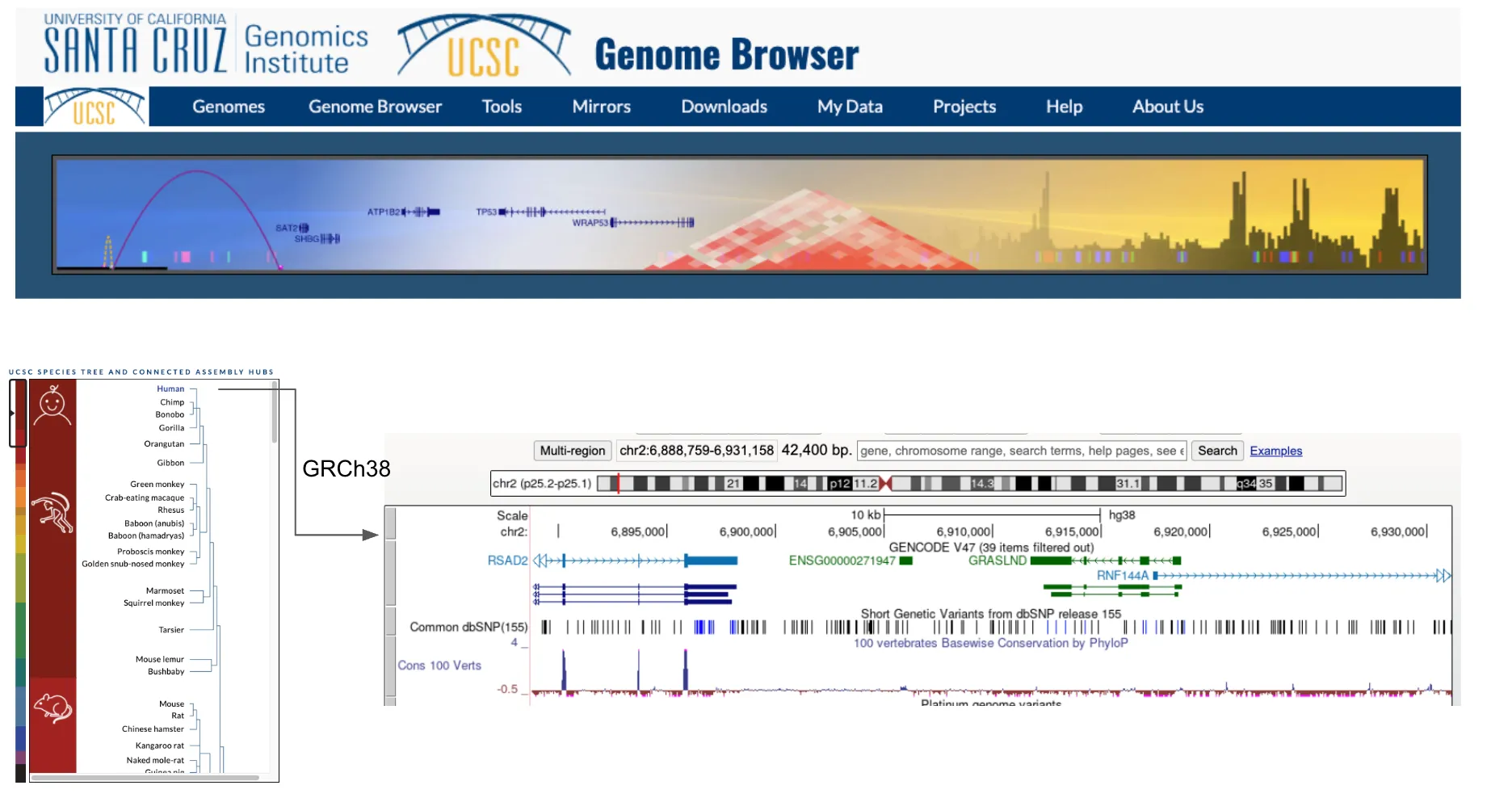
2. Supported data types
Section titled “2. Supported data types”| Data Type | Description |
|---|---|
| bam/cram | Compressed Sequence Alignment/Map tracks |
| bigBed | Item or region tracks |
| bigBarChart | Bar charts of categorical variables displayed over genomic regions |
| bigChain | Genome-wide Pairwise Alignments |
| bigGenePred | Gene Annotations |
| bigInteract | Pairwise interactions |
| bigLolly | Lollipops |
| bigNarrowPeak | Peaks |
| bigMaf | Mulitple Alignments |
| bigPsl | Pairwise Alignments |
| bigWig | Signal graphing tracks |
| hic: Hi-C | Contact matrices |
| halSnake | HAL Snake Format |
| vcfTabix | Variant Call Format |
| vcfPhasedTrio | Variant Call Format Trios |
3. Commonly used data types in Zlab
Section titled “3. Commonly used data types in Zlab”-
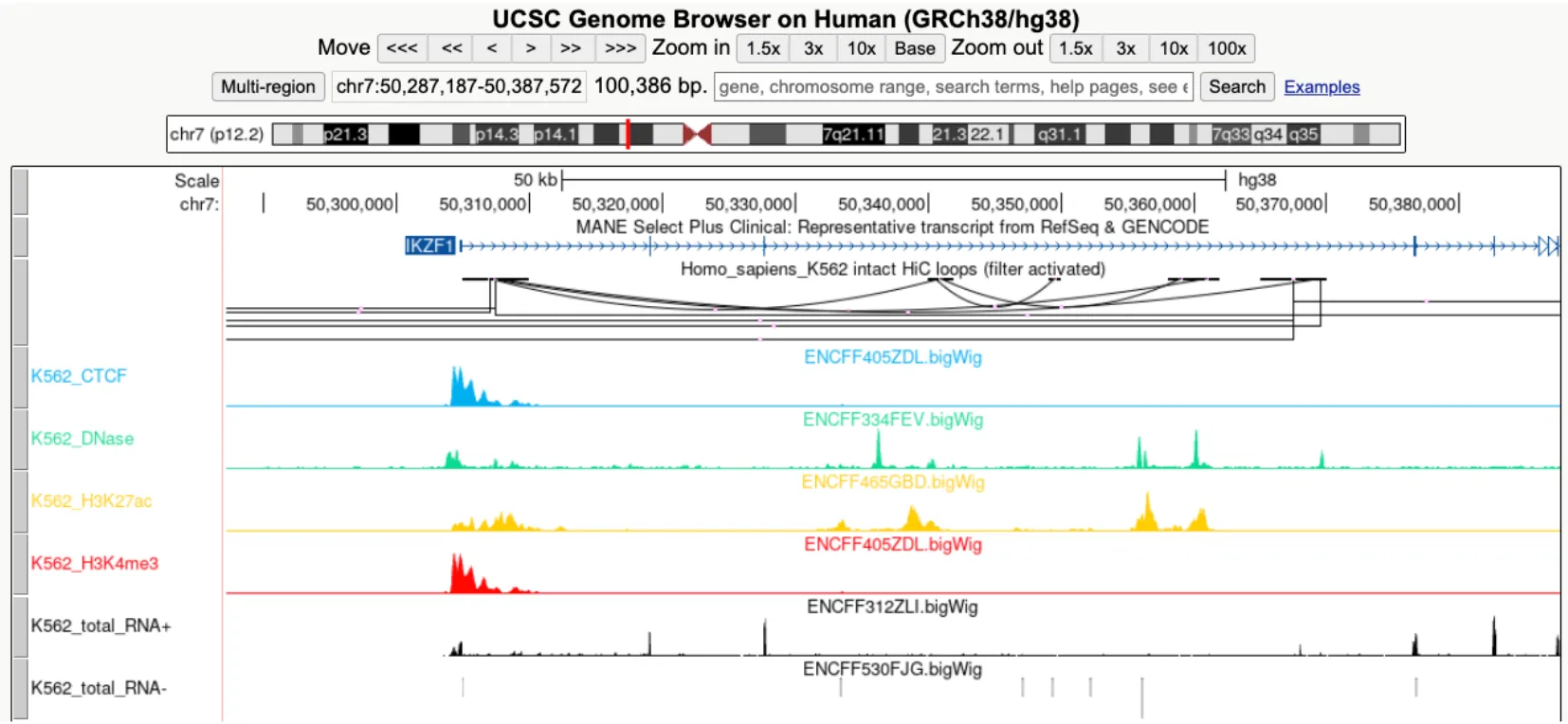
bigBed: Visualize regions such as varians, repeats, cCREs and ChIP-seq peaks.
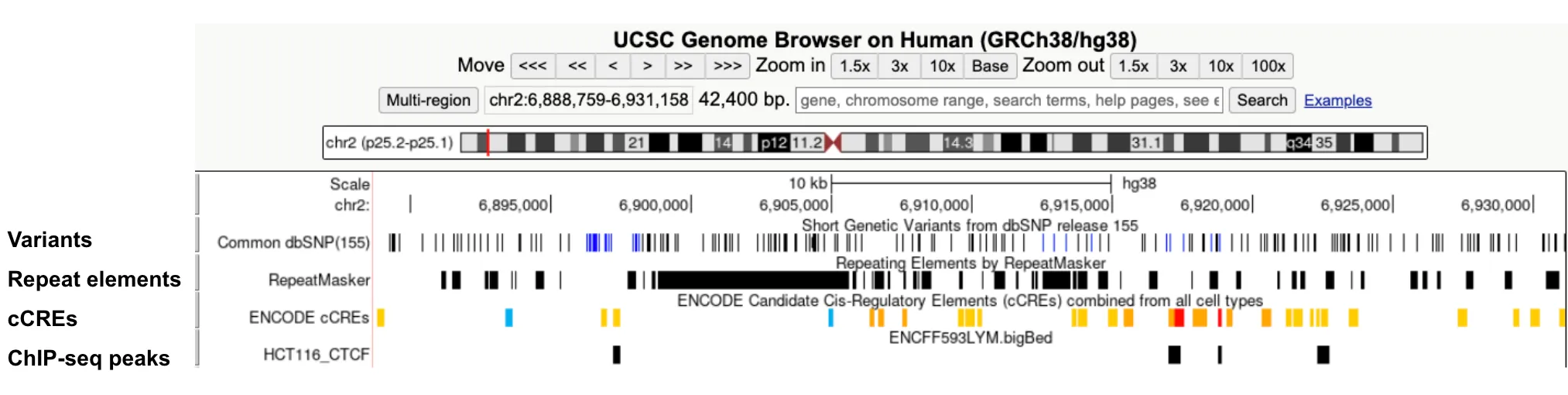
-
bigWig: Visualize signals for assays such as ChIP-seq, ATAC-seq, DNase-seq and RNA-seq.
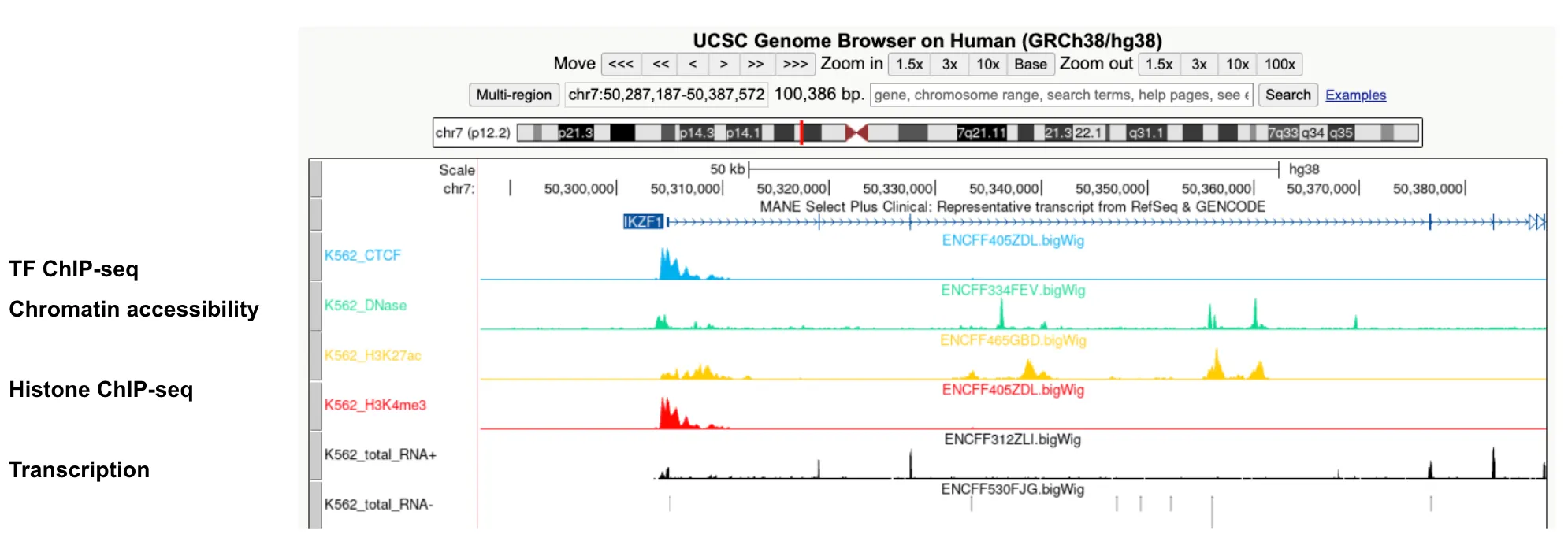
-
bigInteract: Visualize pairwise interactions for assays such as HiC and ChIA-PET.
4. Data preparation
Section titled “4. Data preparation”ENCODE data
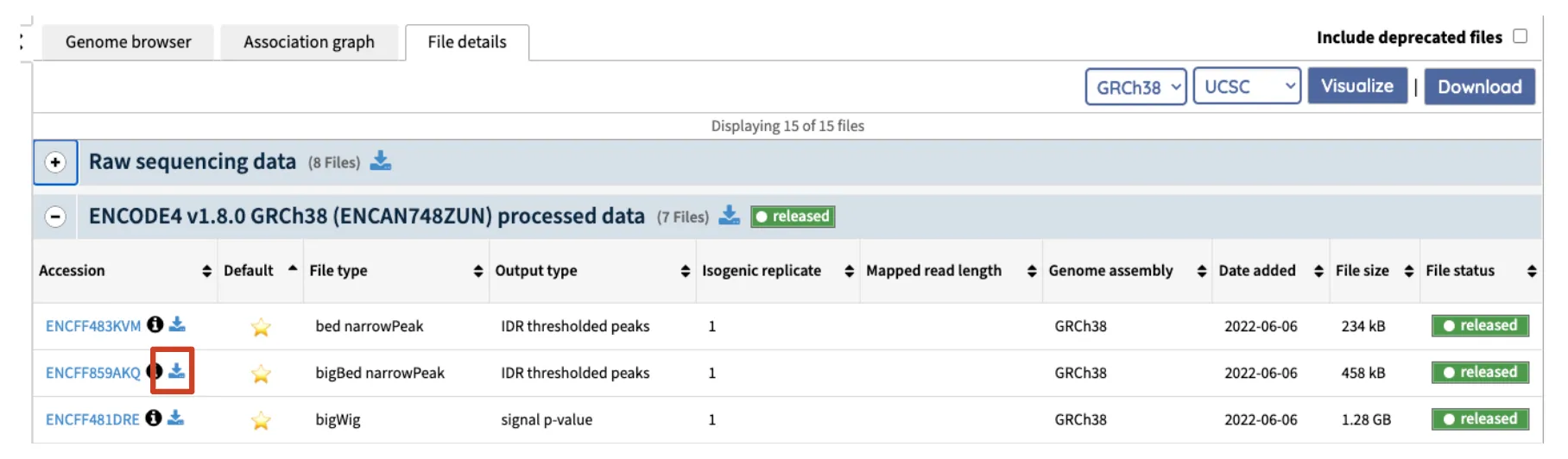
Section titled “ENCODE data”Find the data you want to visualize on the ENCODE portal, right click the download logo and copy the data URL(copy link address) for Genome Browser visualization.
Custom data transformation
Section titled “Custom data transformation”UCSC developed various tools for data transformation.
bigBed
Section titled “bigBed”BED (Browser Extensible Data) format
graph LR A[bed] --> B[bedToBigBed] B --> C[bigBed]
bigWig
Section titled “bigWig”bedGraph format
graph LR A[bedGraph] --> B[bedGraphToBigWig] B --> C[bigWig]
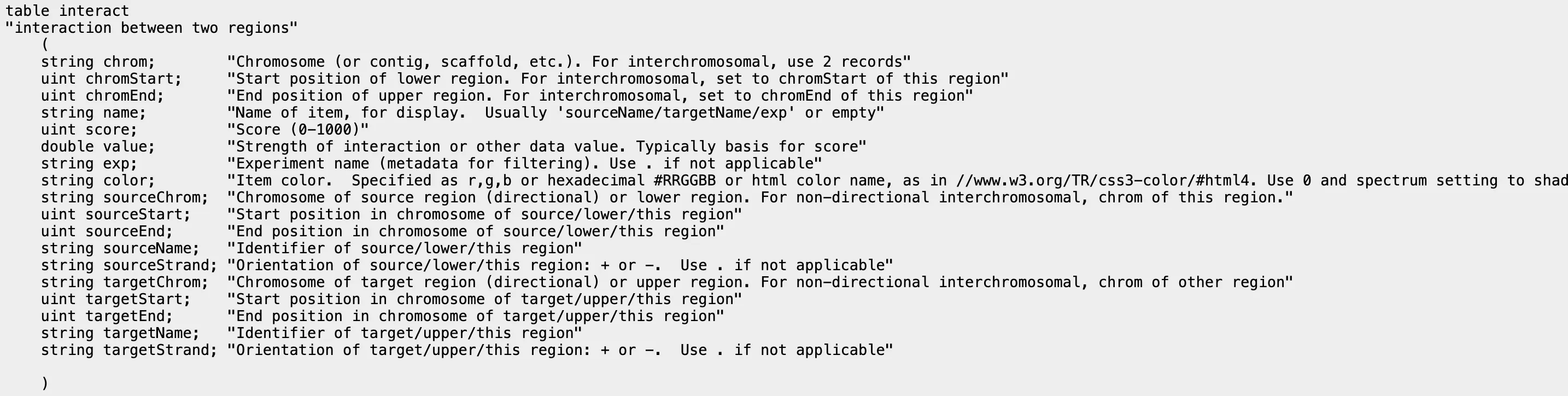
bigInteract
Section titled “bigInteract”bedpe format, bigInteract format
graph LR A[bedpe] --> B[bed5+13] B --> C[bedToBigBed] C --> D[bigInteract]

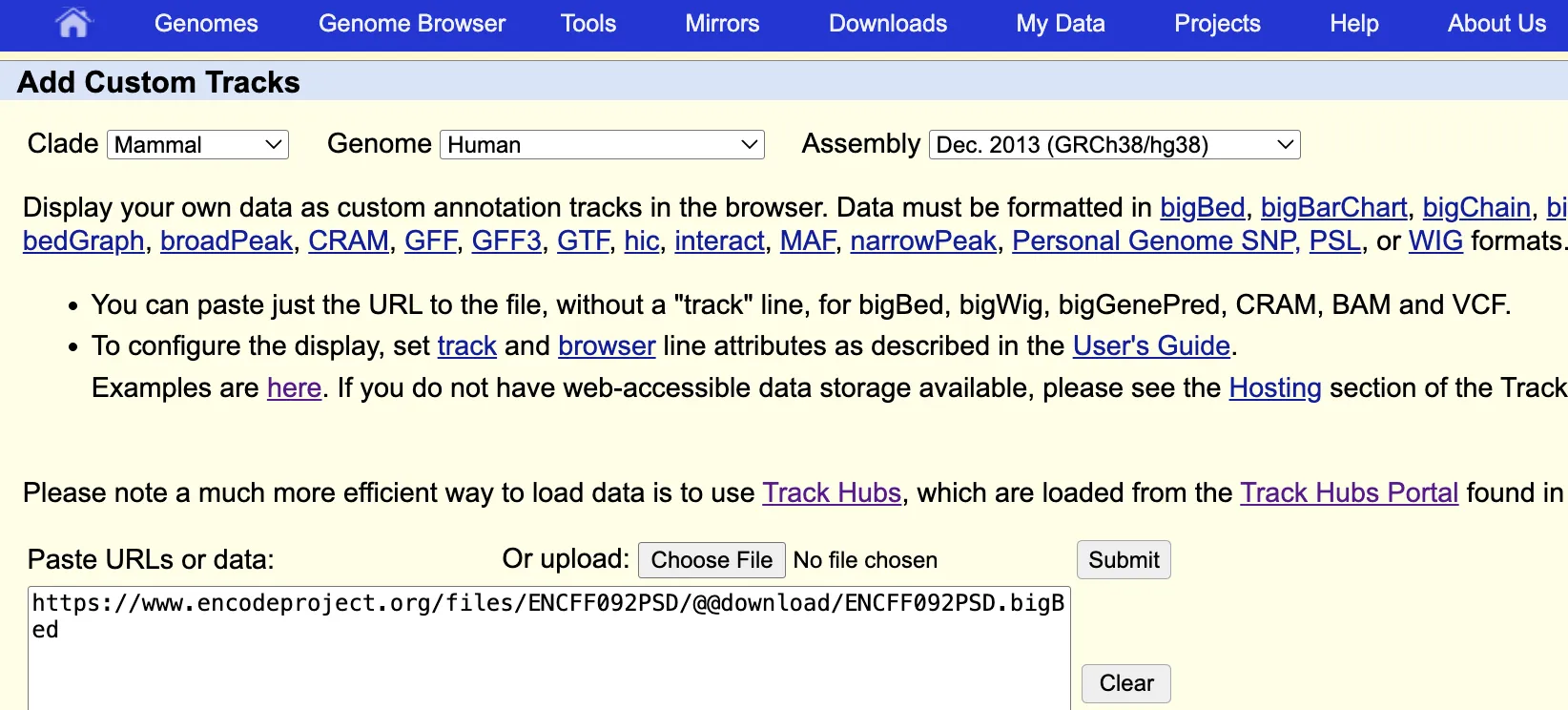
5. How to make custom track
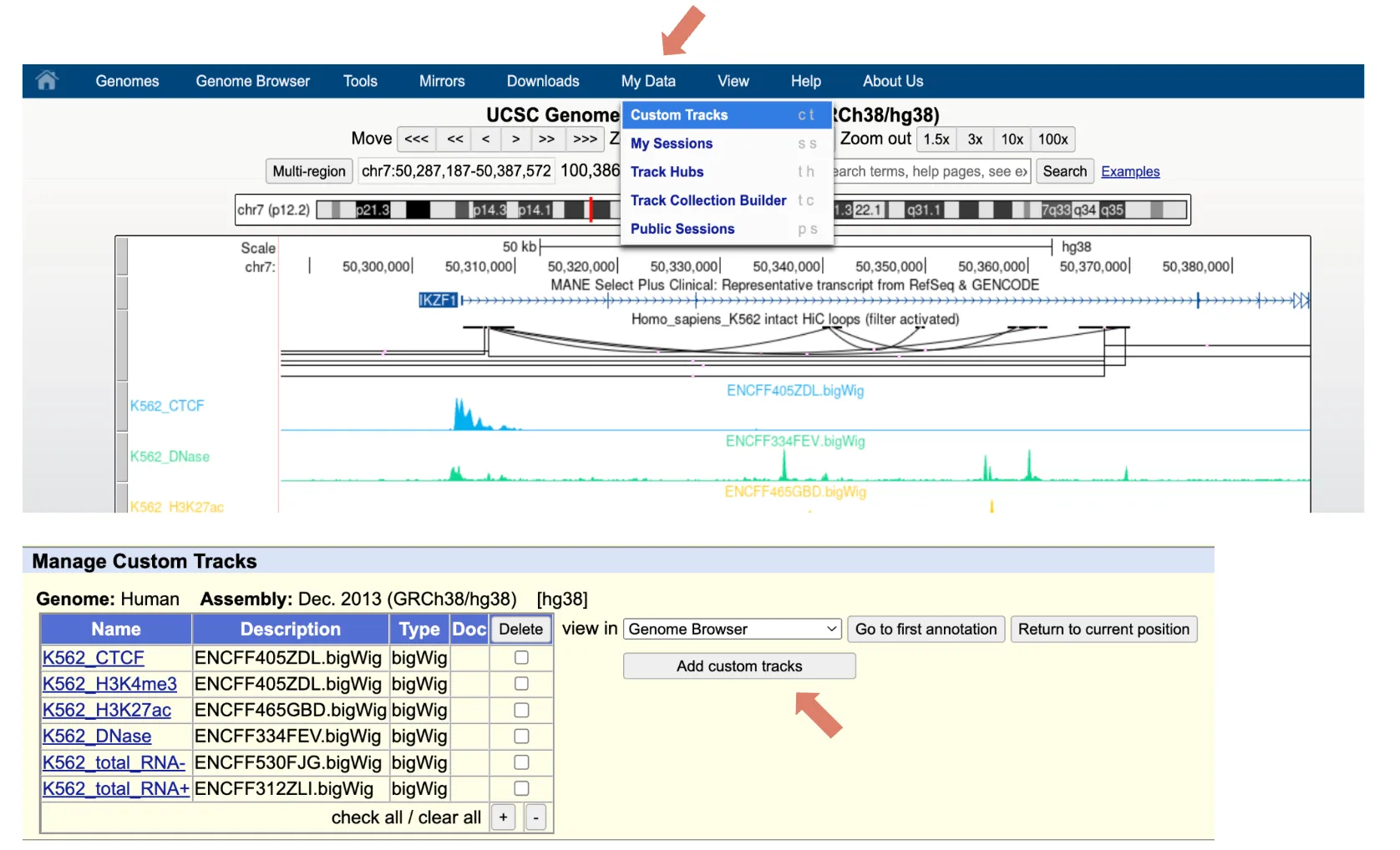
Section titled “5. How to make custom track”Custom track is useful for quickly browse one or a few datasets.
To add custom track, click Custom Tracks under the My Data tab and then click Add custom tracks. In the page, paste in the data URL or upload from local.
6. How to make track hub
Section titled “6. How to make track hub”Track hubs enable quick and organized visualization of large datasets. They also make it easy to share data, since collaborators can view your tracks directly in the genome browser with a simple URL.
Public directory on Zerver
Section titled “Public directory on Zerver”/zata/public_html_users/username is available as https://users.wenglab.org/username (eg. https://users.wenglab.org/gaomingshi).
Set up a folder for track hub
Section titled “Set up a folder for track hub”-
Classic 3-file style (useful if you have data from more than 1 genome assembly)
- hub.txt (description of the track hub)
- genomes.txt (specify the genome assemblies and corresponding trackDb.txt)
- trackDb.txt (define the tracks)
track hub directory setup example:

- 1-file style (easy setup for data from the same genome assembly, eg, GRCh38
- hub.txt (add useOneFile on and genome xxx )
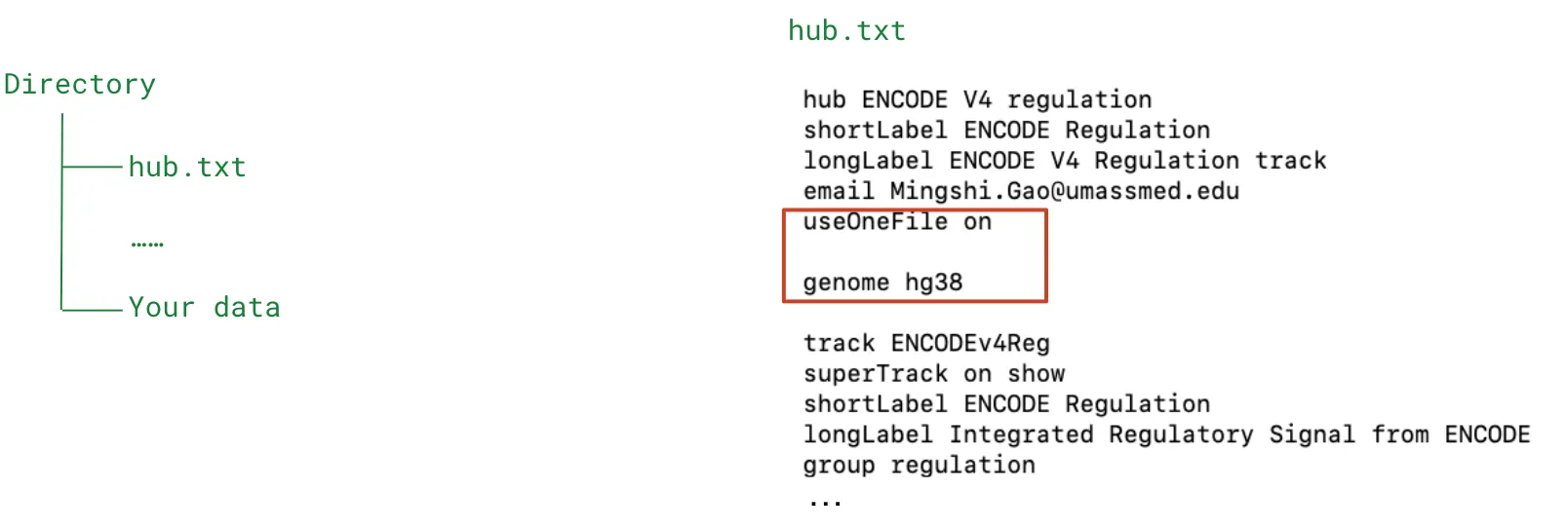
Entries of trackDb.txt
Section titled “Entries of trackDb.txt”Specify each track in the trackDb.txt.

5 basic parameters needed for a track are:
track(track id, needs to be unique)type(data type)bigDataUrl(the path of data, supports both local path and URL)- local file (eg. ENCFF122BJN.bigWig, if the file is in the same directory as trackDb.txt)
- URLs (https://www.encodeproject.org/files/ENCFF122BJN/@@download/ENCFF122BJN.bigWig)
short label(label that shows up at the right side of the track in genome browser view)long label(label that shows up above the track in genome browser view)
other useful parameters:
autoScale On(useful for scaling bigWig view)maxHeightPixels(height of track)negateValues On(useful for set - strand RNA-seq signal as negative value)color(set track color, supports RGB)visibility(set each track to ideal visibility)
Prepare trackDb.txt / one-file hub.txt
Section titled “Prepare trackDb.txt / one-file hub.txt”To add an entry for each dataset, it’s helpful to use a loop to modify a template file by inserting each dataset’s unique metadata. Below is an example of how I created the ENCODE Epigenetics Collection hub.
- Screenshot of ATAC-seq metadata file:

- Template file track.txt:
track File_Type_FIDtype bigWigshortLabel EIDlongLabel NAME File_Type signalbigDataUrl https://www.encodeproject.org/files/FID/@@download/FID.bigWigvisibility fullmaxHeightPixels 50color RGBautoScale on- Code to prepare trackDb.txt:
rm -f trackDb.txtfor File_Type in ATAC DNase CTCF H3K4me3 H3K27acdo cat ${File_Type}-List.txt | while IFS=$'\t' read -r EID FID name BIOTYPE Organ do NAME=$(echo $name | awk -F "_ENCDO" '{print $1}') RGB=$(grep -wi "${File_Type}" signal.color.txt | cut -f 2) echo -e "${File_Type}\t${OID}\t${EID}\t${BIOTYPE}\t${RGB}"
sed -e "s/EID/${EID}/g" -e "s/FID/${FID}/g" \ -e "s/File_Type/${File_Type}/" -e "s/RGB/${RGB}/" \ -e "s|NAME|${NAME}|" track.txt > trackDb.txt donedoneShare track hub with collaborators
Section titled “Share track hub with collaborators”- Share the URL of hub.txt for people to load at track hub page, eg. http://users.wenglab.org/gaomingshi/ENCODE_Reg/hub.txt
- Create a session and share session URL (for advanced users, need to create a UCSC Genome Browser account) see the following links for more details: https://genome.ucsc.edu/cgi-bin/hgSession https://genome.ucsc.edu/goldenPath/help/hgSessionHelp.html
Useful links
Section titled “Useful links”- Hub Track Database Definition
- Public Track Hub’s TrackDb files on Github (Example: ENCODE regulation track hub)
- Lab Custom Track Color Google sheet
- Kaili’s slides on downloading ENCODE data in batch
- My ENCODE metadata tutorial Google colab
Debugging
Section titled “Debugging”shortLabelneeds to be shorter than 17 characters,trackDb.txthas trouble processing ’ (quotation mark). Avoid using it if you can.